
During drug development, you want to know the affinity between a molecule (potential drug) and its target, such as a protein on the surface of a cell. That is: does this particular molecule tightly fit at a specific place and stay there? Key in a lock, hand in a glove, are the analogies.
Today ML can't solve affinity
Machine learning (ML) and deep learning should be good at estimating affinity, but in the general case, they are not. The techniques today don't generalise (the "out of distribution" performance is poor), and there's not enough data in the first place.
This is bad because you want a fast estimate of this affinity to know which potential drugs are worth exploring further. (Techniques like FEP are rigorous, but slow, as they simulate details of the physics).
ML vs physics-inspired scoring models
It was great to chat with smart people about this at the UCL/Structural Genomics Consortium meeting on ML and drug discovery back in October 2025. I learned a lot, as the idiot in the room, and a few things stuck with me.
One was Fergus Imrie's talk on OpenBind, the UK initiative to generate more quality affinity data from real-world experiments with molecules and targets.
In particular, he had a nice experiment comparing ML predictions of affinity against physics-inspired scoring models ("docking tools"—not comparable to true physics, but much faster).
It went something like this: Given that we have an idea for how molecules bind to their targets, if you perturb a "good" molecule, you'd expect a model's score to go down.
You do see that decrease for physics-based tools, which work fast by taking shortcuts to "score" an interaction. But for deep-learning-based models, relying on patterns from the data, the change is all over the place:
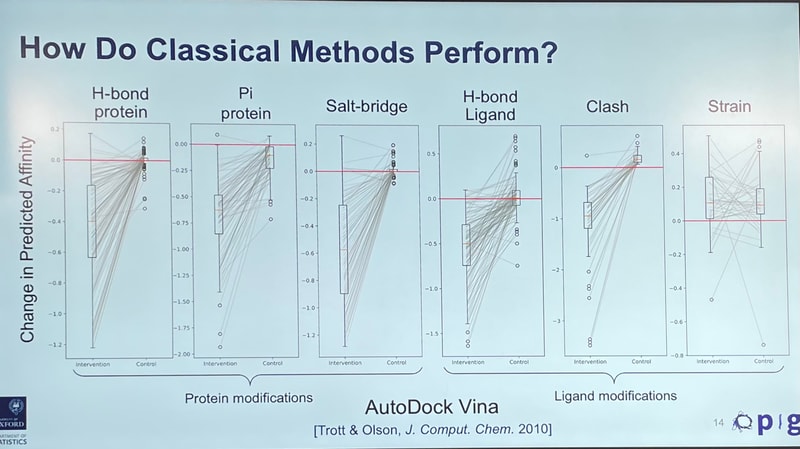
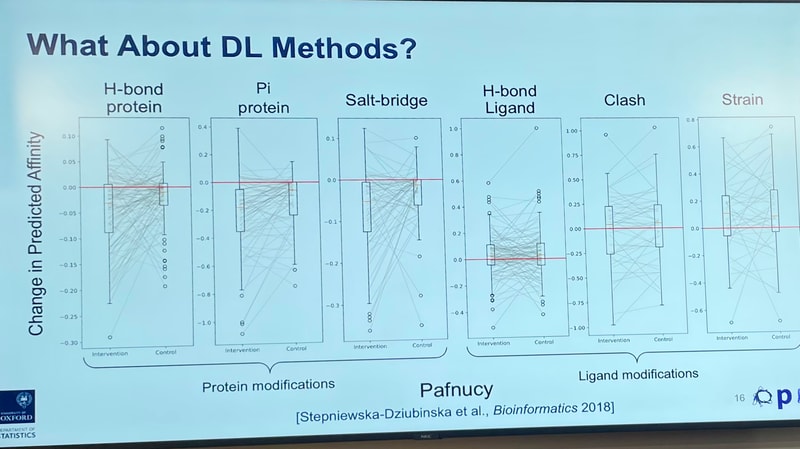
We have to wait for the paper to understand this properly, but it's a lovely experiment.
What to do?
The patterns ML systems are learning don't appear to capture the underlying reality, despite "magical sounding claims".
The answer being pursued, and the focus of the meeting, is to gather more data, with higher quality. We're going to need a lot of it.
—
