
Yesterday I was at UCL for a presentation called “Target 2035: A 2000-protein experiment to ligand the human proteome”. It’s a project from the Structural Genomics Consortium (SGC, which UCL has just joined).
The aim is to create large and open dataset for drug discovery
Thinking of the success of AlphaFold in predicting protein folding, that depended on a high quality curated dataset. An analogous resource doesn’t exist for small molecules (or “drugs” to you and me).
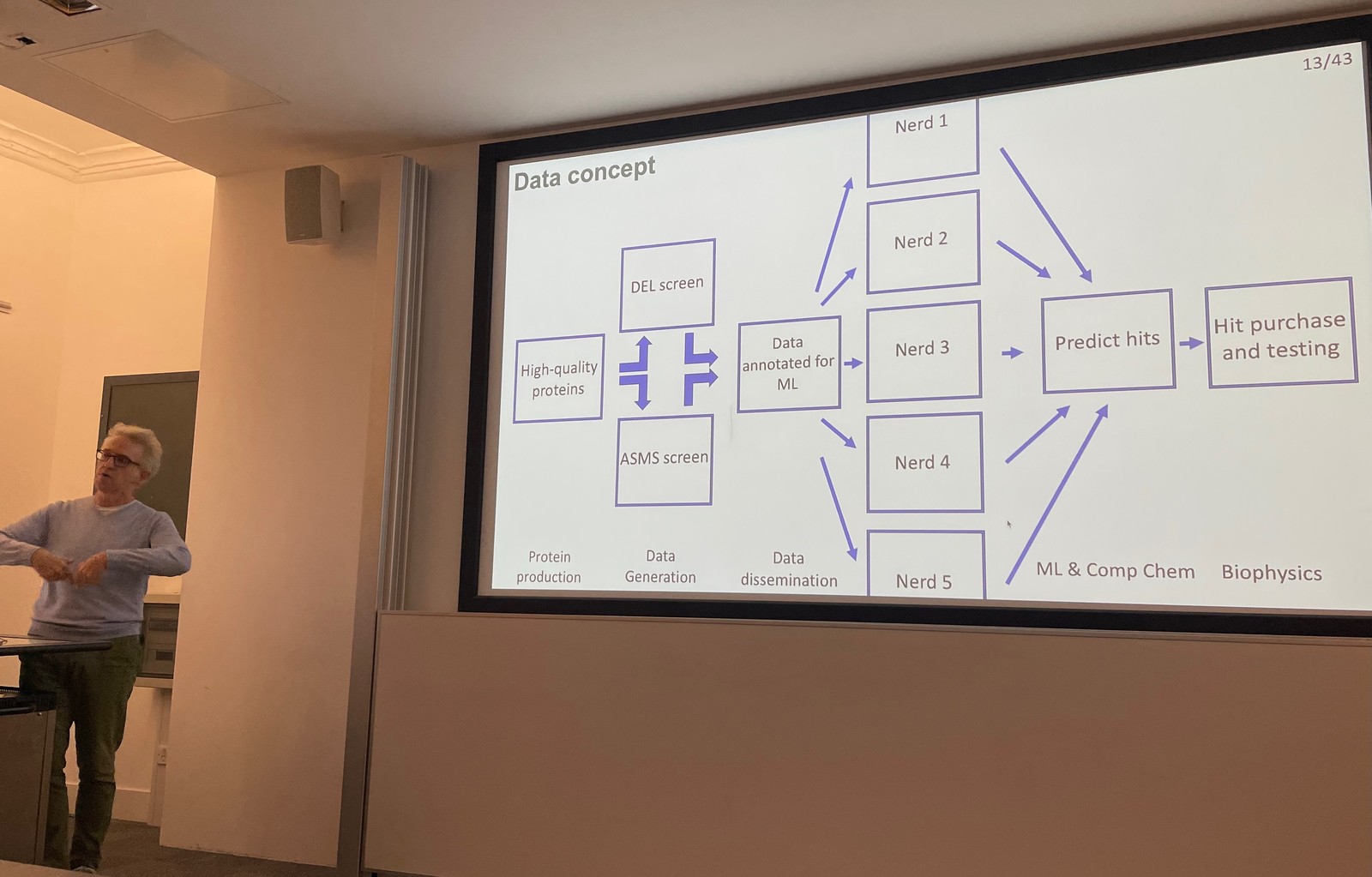
SGC's plan is to fix this by large scale screening of proteins, producing a data set of molecules that bind to those proteins.
To break that down, very roughly, small molecules “keys” interact with protein "locks” on the surface of a cell (let’s say) to trigger some kind of effect (maybe we’d call that a “treatment”). If you want to predict what an arbitrary molecule might be able to do, you need to collect data on lots of proteins and lots of molecules to know what does and doesn’t “bind” to a protein.
The plan, then, is to collect information on 10 billion molecules per protein (sounds like a lot), and to do that for many proteins. This data will be open, which might well help drive advances in machine learning. That could be using ML to generate novel binders for a protein of interest, or be able to filter a list of molecules quickly to find likely binders.
DNA-encoded libraries as a source of truthiness
The data has to come from actual experiments with the physical molecules and proteins. SGC has a couple of ways to do that, and one is DEL (DNA-encoded libraries).
My understanding of DEL is limited. As I understand it, organisations have found a way to build up molecules, step-by-step, and at each step attach a piece of DNA to the molecule. The DNA “tail” in effect describes the synthesis steps taken to generate the molecule.
With that, you mix the molecules with copies of your protein, and shortly after kind of wash it away. What you’re left with is just the molecules that have bound to the protein.
Finally, read the DNA, and you know what those remaining molecules are (or at least the steps to get to the molecule). These "hits” don’t tell you what the molecule does, but it tells you it has a chance of doing something with the protein.
That’s useful in itself, but the next step is to use ML to learn the patterns from that data. It goes under the banner of “DEL-ML”.
This is exciting stuff
This should produce a ton of data, and hopefully lead to AlphaFold-levels of advancement for small molecules.
Part of the secret sauce here is the quality of the data generated. I thought it interesting that a representative from Pfizer noted that they’d prefer to contribute to— and use—a high quality open dataset over than their own historic data sets they have accumulated over the last 175 years.
* * *
Links for me to follow up
- The future of machine learning for small-molecule drug discovery will be driven by data, Nature, 15 October 2024.
- For chemists, the AI revolution has yet to happen, Nature, 17 May 2023.
- Machine Learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding, Journal of Medicinal Chemistry, 11 June 2020.
- A resource to enable chemical biology and drug discovery of WDR Proteins, bioRxiv, 4 March 2024.
- A data science roadmap for open science organizations engaged in early-stage drug discovery, Nature Communications, 5 July 2024.