
One step in drug discovery is a claim on intellectual property. There are various form of this, including rights on data from experiments, but a patent on “composition of matter” is an obvious one.
This means, if you’re generating molecules, it would be useful to know if the molecule is subject to a patent—although not as useful as you might think, which I’ll return to at the end.
I’ve explored how to do this computationally, and this post is a write up and code of that experience. The summary is: read the molecules as text, store them in a Bloom filter (a fixed size fast yes/no look up), and you’re done. It’s an order of magnitude faster than looking up a molecule in a database.
Data
SureChEMBL is an open source collection of 23 million compounds (a splash in the ocean), mined from patents.
I picked up the “MAP files” which contains a description of the molecules and the patents they related to. The format for a molecule is SMILES, which we don’t need to go into, but know that it is a text representation of a molecule.
Getting the quarterly dump of the data is easy enough:
brew install lftp
lftp -c 'mirror \
ftp://ftp.ebi.ac.uk/pub/databases/chembl/SureChEMBL/data/map'
That gives you a directory of .txt.gz tab-separated files. The molecule is column 2:
gzip -cd *.txt.gz | cut -d$'\t' -f 2 > smiles.txt
There are 373,616,491 entries, but after you dedupe that you have 23,465,171 molecules.
The basic idea
Bloom filters are magic:
- You have a fixed array of bits, starting with all zeros.
- To “store” an item, you compute a numeric hash for the item and that gives you the index in your array to set to 1. (In reality, you apply a few hash functions, meaning you set more than one bit.)
- To “look up” an item, you compute the hash and see if all the bits are 1. If they are, you’ve seen the item before; otherwise you haven’t.
This is nice because it’s a fixed size and fast.
However, if you store lots of items in the filter all the bits will be eventually be 1—meaning you get 100% false positives. That’s manageable via parameters: you can trade off the size of the array against the false positive rate.
Applying this to molecules is trivial, especially as it’s been done before: Medina & White, Bloom filters for molecules, Journal of Cheminformatics, 2023. That work was for checking if you can buy a molecule, but it’s trivial to switch it to checking if someone has patented a molecule.
The code
I didn’t re-implement the C and Python code in that paper, but wrapped a command line around the Rust fastbloom library (which implements ideas from “Less Hashing, Same Performance: Building a Better Bloom Filter”).
It uses the SipHash-1-3 hasher by default — it’s not clear to me the benefit of various hashers, but experimentally this works and is efficient.
My code is at: https://github.com/d6y/molbloom. Thanks to all the hard work being done in a library, I only had to write ~100 lines of code.
The code has two commands: build to construct a Bloom filter from text (one item per line); and query to answer true or false to a list of items.
The slow part of it is serialising the filter to disk. It’s pretty big. The approximate formula for computing the size you need for a given false positive rate tells me to store 23 million molecules with 1% chance of a false positive I need an array of 27.5 million bytes (26M).
Testing
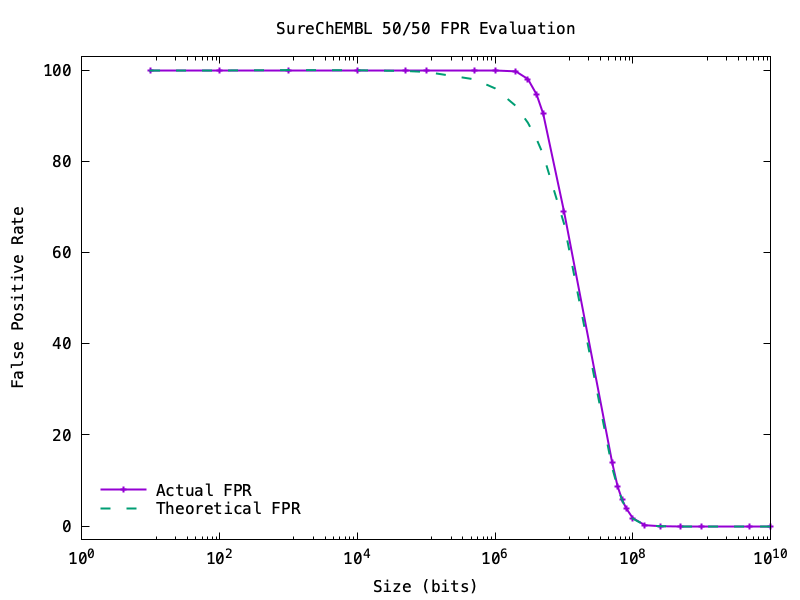
Does this work? Following the example from Bloom filters for molecules, I split the data into half. The first half I used to build the filter, and the second half to test the filter. The test should be all false (0% false positives).
You can see this work as you vary the size of the Bloom filter:
Gotchas
This implementation doesn’t care about what text you give it. That means you need to be careful to normalise any query to be consistent with the data used to build the hash.
SureChEMBL data is:
- SMILES: “ChemAxon canonical kekule-based SMILES representation”
- InChiKey: “ChemAxon-generated standard InChI key” — this is another text representation of a molecule.
It works, how useful is this?
It’s not as useful as it feels like it should be:
- There’s the false positive rate, but we’ve seen we can manage that.
- Coverage isn’t 100%: it is estimated that around 60% of patented molecules are in SureChEMBL — that’s a good percent, but it’s not definitive.
- Then there are Markush structures, a template used in patents for molecules with substitution points. Enumerating a pattern like that could give you millions of molecules, and they are not in SureChEMBL.
- Even if the molecules is subject to a patent, you don’t know if it’s relevant or if it’s expired. Deals are always possible, I assume.
- You don’t know if your molecule is potentially patentable (is it a nature product? Is there a non-obvious step?)
In summary:
- Getting a
true(probably known to SureChEMBL) from the code doesn’t mean it is subject to a valid patent. - Getting a
false(not in SureChEMBL) doesn’t mean it’s not covered by a relevant patent.
This is something to treat as an indication, a flag, to feed in to a prioritisation exercise.